
Study: Some catalysts contribute their own oxygen for reactions
Chemical reactions that release oxygen in the presence of a catalyst, known as oxygen-evolution reactions, are a crucial part of chemical energy storage processes, including water splitting, electrochemical carbon dioxide reduction, and ammonia production. The kinetics of this type of reaction are generally slow, but compounds called metal oxides can have catalytic activities that vary over several orders of magnitude, with some exhibiting the highest such rates reported to date. The physical origins of these observed catalytic activities is not well-understood.
Now, a team at MIT has shown that in some of these catalysts oxygen doesn’t come only from the water molecules surrounding the catalyst material; some of it comes from within the crystal lattice of the catalyst material itself. The new findings are being reported this week in the journal Nature Chemistry, in a paper by recent MIT graduate Binghong Han PhD ’16, postdoc Alexis Grimaud, Yang Shao-Horn, the W.M. Keck Professor of Energy, and six others.
The research was aimed at studying how water molecules are split to generate oxygen molecules and what factors limit the reaction rate, Grimaud says. Increasing those reaction rates could lead to more efficient energy storage and retrieval, for example, so determining just where the bottlenecks may be in the reaction is an important step toward such improvements.
The catalysts used to foster the reactions are typically metal oxides, and the team wanted “to be able to explain the activity of the sites [on the surface of the catalyst] that split the water,” Grimaud says.
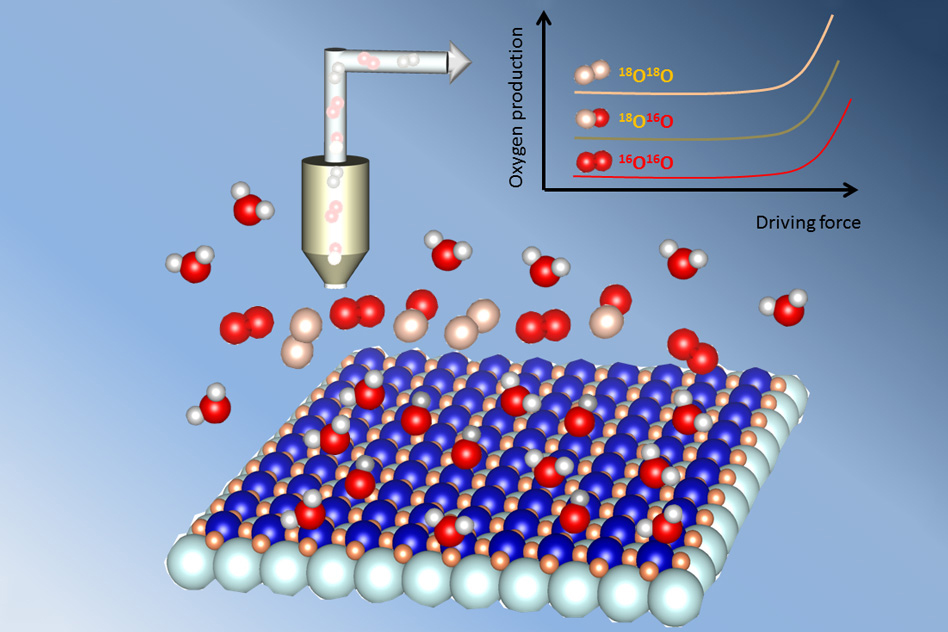
The question of whether some oxygen gets stored within the crystal structure of the catalyst and then contributes to the overall oxygen output has been debated before, but previous work had never been able to resolve the issue. Most researchers had assumed that only the active sites on the surface of the material were taking any part in the reaction. But this team found a way of directly quantifying the contribution that might be coming from within the bulk of the catalyst material, and showed clearly that this was an important part of the reaction.
They used a special “labeled” form of oxygen, the isotope oxygen-18, which makes up only a tiny fraction of the oxygen in ordinary water. By collaborating with Oscar Diaz-Morales and Marc T. Koper at Leiden University in the Netherlands, they first exposed the catalyst to water made almost entirely of oxygen-18, and then placed the catalyst in normal water (which contains the more common oxygen-16).
Upon testing the oxygen output from the reaction, using a mass spectrometer that can directly measure the different isotopes based on their atomic weight, they showed that a substantial amount of oxygen-18, which cannot be accounted for by a surface-only mechanism, was indeed being released. The measurements were tricky to carry out, so the work has taken some time to complete. Diaz-Morales “did many experiments using the mass spectrometer to detect the kind of oxygen that was evolved from the water,” says Shao-Horn, who has joint appointments in the departments of Mechanical Engineering and Materials Science and Engineering, and is a co-director of the MIT Energy Initiative’s Center for Energy Storage.
With that knowledge and with detailed theoretical calculations showing how the reaction takes place, the researchers say they can now explore ways of tuning the electronic structure of these metal-oxide materials to increase the reaction rate.
The amount of oxygen contributed by the catalyst material varies considerably depending on the exact chemistry or electronic structure of the catalyst, the team found. Oxides of different metal ions on the perovskite structure showed greater or lesser effects, or even none at all. In terms of the amount of oxygen output that is coming from within the bulk of the catalyst, “you observe a well-defined signal of the labeled oxygen,” Shao-Horn says.
One unexpected finding was that varying the acidity or alkalinity of the water made a big difference to the reaction kinetics. Increasing the water’s pH enhances the rate of oxygen evolution in the catalytic process, Han says.
These two previously unidentified effects, the participation of the bulk material in the reaction, and the influence of the pH level on the reaction rate, which were found only for oxides with record high catalytic activity, “cannot be explained by the traditional mechanism” used to explain oxygen evolution reaction kinetics, Diaz-Morales says. “We have proposed different mechanisms to account for these effects, which requires further experimental and computational studies.”
“I find it very interesting that the lattice oxygen can take part in the oxygen evolution reactions,” says Ib Chorkendorff, a professor of physics at the Technical University of Denmark, who was not involved in this work. “We used to think that all these basic electrochemical reactions, related to proton membrane fuel cells and electrolyzers, are all taking place at the surface,” but this work shows that “the oxygen sitting inside the catalyst is also taking part in the reaction.”
These findings, he says, “challenge the common way of thinking and may lead us down new alleys, finding new and more efficient catalysts.”
The team also included Wesley Hong PhD ’16, former postdoc Yueh-Lin Lee, research scientist Livia Giordano in the Department of Mechanical Engineering, Kelsey Stoerzinger PhD ’16, and Marc Koper of the Leiden Institute of Chemistry, in the Netherlands. The work was supported by the Skoltech Center for Electrochemical Energy, the Singapore-MIT Alliance for Research and Technology, the Department of Energy, and the National Energy Technology Laboratory.


